Benchmarking variant detection
in viral populations
Interactive figures for the analysis presented in
McCrone JT and Lauring AS, J. Virol. 90(15):6884, 2016
Interactive figures for the analysis presented in
McCrone JT and Lauring AS, J. Virol. 90(15):6884, 2016
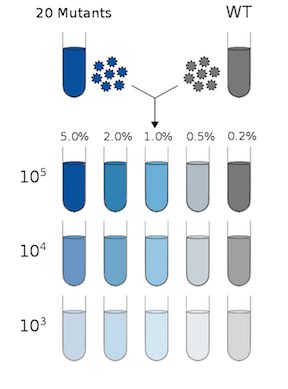
Advances in sequencing technology have made it feasible to sequence patient-derived viral samples at a level sufficient for detection of rare mutations. These high-throughput, cost-effective methods are revolutionizing the study of within-host viral diversity. However, these techniques are error prone, and the methods commonly used to control for these errors have not been validated under the conditions that characterize patient-derived samples. In our study we showed that these conditions affect measurements of viral diversity. We found that the accuracy of previously benchmarked analysis pipelines were greatly reduced under patient-derived conditions. By sequencing known mixtures of single mutants we were able to identify biases in our method and improve our accuracy to acceptable levels. Here, we provide a "light", interactive version of the main figure so others may explore the impact different quality thresholds have on the accuracy of the data. A fuller shiny application can be downloaded below and provides more options for variant identification.
If you enjoyed this interactive figure and would like to explore the data more
be sure to check out the full Shiny app.